After the Miracle Drug
Cystic fibrosis once guaranteed an early death—but a medical breakthrough has given many patients a chance to live decades longer than expected. What do they do now?

Photographs by Fumi Nagasaka
They call it the Purge.
You have experienced, in a modest way, something like it in the waning days of a bad cold, when your lungs finally expel their accumulated gunk. The rattle in your chest quiets. Your sinuses clear. You smell again: the animal sweetness of your children’s hair, the metallic breeze stirring a late-summer night. Your body, which oozed and groaned under the yoke of illness, is now a perfectly humming machine. Living is easy—everything is easy. How wonderful it is to breathe, simply breathe.
Imagine, though, that you had never been able to simply breathe. Imagine that mucus—thick, copious, dark—had been accumulating since the moment you were born, thwarting air and trapping microbes to fester inside your lungs. That you spent an hour each day physically pounding the mucus out of your airways, but even then, your lung function would spiral only downward, in what amounted to a long, slow asphyxiation. This was what it once meant to be born with cystic fibrosis.
Then, in the fall of 2019, a new triple combination of drugs began making its way into the hands of people with the genetic disease. Trikafta corrects the misshapen protein that causes cystic fibrosis; this molecular tweak thins mucus in the lungs so it can be coughed up easily. In a matter of hours, patients who took it began to cough—and cough and cough and cough in what they later started calling the Purge. They hacked up at work, at home, in their car, in bed at night. It’s not that they were sick; if anything, it was the opposite: They were becoming well. In the days that followed, their lungs were cleansed of a tarlike mucus, and the small tasks of daily life that had been so difficult became unthinkingly easy. They ran up the stairs. They ran after their kids. They ran 10Ks. They ran marathons.
Cystic fibrosis once all but guaranteed an early death. When the disease was first identified, in the 1930s, most babies born with CF died in infancy. The next decades were a grind of incremental medical progress: A child born with CF in the ’50s could expect to live until age 5. In the ’70s, age 10. In the early 2000s, age 35. With Trikafta came a quantum leap. Today, those who begin taking the drug in early adolescence, a recent study projected, can expect to survive to age 82.5—an essentially normal life span.
CF was one of the first diseases to be traced to a specific gene, and Trikafta is one of the first drugs designed for a specific, inherited mutation. It is not a cure, and it doesn’t work for all patients. But a substantial majority of the 40,000 Americans with CF have now lived through a miracle—a thrilling but disorienting miracle. Where they once prepared for death, they now have to prepare for life. “It’s like the opposite of a terminal diagnosis,” Jenny Livingston told me.
Jenny spent her 20s in and out of the hospital for CF-related lung infections. During her frequent weeks-long stays, she made some of her best friends in the CF ward, only to watch them succumb, one by one, to the disease that she knew would eventually kill her too. More than anything, she hoped to live long enough to see her daughter graduate from high school.
[From the December 2020 issue: Sarah Zhang on the last children of Down syndrome]
Today, Jenny is 36. Four years into taking Trikafta, she’s the healthiest she’s been in her adult life. Her daughter is 14, a lanky high-school freshman. They’re both obsessed with Harry Styles, and after Jenny started on Trikafta, they flew together to see him live—twice. They learned to hunt deer with Jenny’s partner, Randy. They often go up into the aspen- and fir-topped mountains that overlook their little town in central Utah. Jenny’s last hospitalization—four years ago, just before she started Trikafta—is now more distant in time than her daughter’s future graduation.
Having lived one life defined by cystic fibrosis, Jenny wonders: What is she going to do with her second life?
Jenny was born in 1987, the youngest of her parents’ five children together and the third to have cystic fibrosis. Given the family history, the doctors knew to test her as an infant, wrapping her forearm in plastic until a sheen of sweat appeared on her skin: the classic “sweat test” for cystic fibrosis. The faulty protein in CF cannot control the balance of salt and water in the body, which results in mucus that is unusually thick and sweat that is unusually salty. In medieval Europe, centuries before anyone understood why, a proverb foretold the fate of children with salt on their skin: “Woe to the child who tastes salty from a kiss on the brow, for he is cursed and soon will die.”
The 1980s, suffice it to say, were not the Middle Ages. By the time Jenny was born, her two older sisters with cystic fibrosis—Shannan, 8, and Teresa, 7—were on a strict schedule of mucus-clearing chest therapy and medications that had kept them alive past toddlerhood. Shannan wasn’t diagnosed until she was 13 months old. “I knew when she was born that there was something wrong,” their mother, Lisa, told me. As a newborn, Shannan projectile vomited and blew out her diapers constantly. When she got older, she was often so insatiably hungry that she would cry when a spoon scraped the bottom of a near-empty food jar. She scarfed down five pancakes at a time. In the baby photos in Lisa’s scrapbook, she is all skinny legs and big, swollen belly—a classic sign of malnutrition.
Shannan was starving, it turned out. Food was passing through her body undigested because her pancreas had been damaged as a result of thick mucus blocking the ducts that release digestive enzymes. Cystic fibrosis was originally named, in fact, for the fibrous cysts that a 1930s pathologist saw in the pancreases of babies who had died. An early epiphany helped doctors overcome the malfunctioning pancreas, though: The missing enzymes could be replaced with pills. By the time of Shannan’s diagnosis, CF was known as a disease of the lungs, in which sticky mucus made fertile ground for bacteria, and the cycle of infection and scarring, infection and scarring would eventually cause the lungs to fail.
Lisa relayed the news of Shannan’s diagnosis over the phone to her husband, Tom, who was at work. As she repeated the doctor’s words, their awful meaning sank in. Their daughter would not live long. They would watch her die. In that moment, the two of them broke down on the phone, the physical distance between them collapsed by grief.
Shannan died when she was 14. “I remember the sound of her oxygen machine more than her voice,” Jenny told me. The rumble and puff of the machine had run in the background of their home, punctuated by chronic coughs from all three girls with CF. But neither Teresa nor Jenny was ever as sick as Shannan was in childhood—due perhaps to chance or to being diagnosed and starting treatments earlier in life. Even when they were newborns, their mother coaxed applesauce sprinkled with enzymes into their mouth, so they could absorb nutrients from their milk.
Not long after Shannan died, Lisa and Tom divorced—their marriage had been strained even before the loss of their daughter—and they both eventually remarried. Despite the upheavals in her family, Jenny remembers her childhood as quite normal. Yes, she had to take the enzymes with every meal, and she had to clear her lungs of mucus every day—first by having her parents pound on her chest and back and later by using an oscillating vest that shook her body. As inhaled CF drugs were developed, they were added to her daily regimen. She went to the hospital for annual preventive “tune-ups,” but she was never sick enough to need emergency hospitalizations, and CF did not seem to hold her back.
Lisa thinks of Jenny as her sassy daughter. Her youngest was always stubborn, always a go-getter. Through the Make-A-Wish Foundation, she was able to get a horse, which she entered in local shows and rode through the foothills just outside town. In the summer, the salt from the dried sweat on her arms became crystals that glimmered in the sun, a subtle reminder of the disease still inside her. The invincibility of youth, however, made her think she had perhaps escaped her oldest sister’s fate.
At 19, Jenny married a local boy she had fallen in love with, and at 21, she was shocked to find herself pregnant: “A very, very happy surprise.” She had always longed to be a mother. As a young girl, she once drew a picture proclaiming that she would grow up to have six children. The drawing “broke my heart,” says her stepmother, Candy. Even if Jenny lived long enough, cystic fibrosis often causes fertility issues—in many women, thickened cervical mucus is thought to prevent pregnancy, and in almost all men, sperm ducts never develop because of blockages that occur in utero. And at the time, doctors often recommended against pregnancy for health reasons.
But Jenny pushed the worries out of her mind. She was simply happy. She set up a crib and painted the nursery. In retrospect, the fevers and shortness of breath she began to feel were not just the normal discomforts of pregnancy, but she didn’t clock it then. She had an uneventful labor, and gave birth to a healthy baby girl. They named her Morgan.
The trouble started in the following months. Six weeks after giving birth, Jenny went back to work. Between nursing and soothing and diapering a newborn, she could no longer keep up her treatment routine. She sometimes also skipped medications when she couldn’t afford them with the pay from her job as a bank teller and her husband’s as a welder.
Then she caught a bug. It was 2009, the year of swine flu, so it could have been that or a more mundane cold, but either way, it triggered something deep in her lungs. She started feeling short of breath. By the time she got to a CF specialist at a hospital two hours away, in Salt Lake City, she could not walk from the car to the front door. She was too weak to stand for her lung-function test. She collapsed into her hospital bed, and for the next several days, she was unable to use the toilet or shower on her own. Convinced that she would die 100 miles from her three-month-old daughter, she had a terrible revelation: “This is why they said ‘Don’t have kids.’ ”
This was Jenny’s first CF pulmonary exacerbation, when lung function plummets from an acute infection. Doctors inserted her first PICC line, a catheter that runs from the upper arm to the heart, delivers antibiotics, and stays in place longer than an IV. She recovered, but just months later, she was back in the hospital with another exacerbation. Then another and another, and on this went for the next several years. Jenny counted for me the PICC-line scars still visible as white dots on each arm—at least 10 on the left, 16 on the right. When the veins in her arms started to reject PICC lines, doctors placed a port under her right collarbone for easy access to her central vein.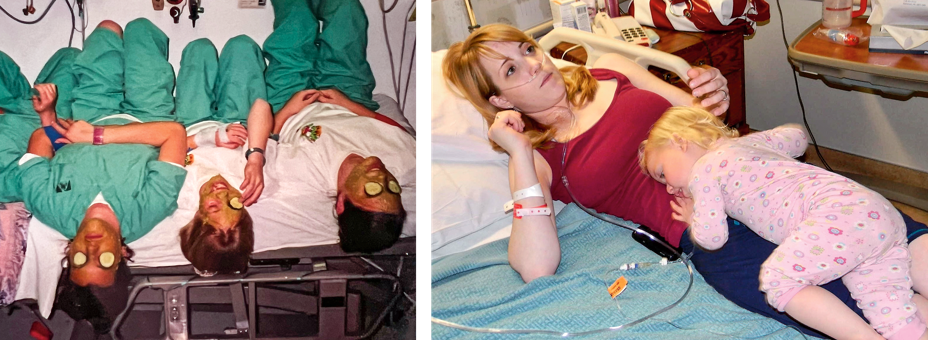
Each infection scarred her lungs; each exacerbation eroded her lung function. The disease that had been a minor plot point in her life became one of its major storylines, and the people in the hospital became recurring characters. At the University of Utah’s CF center, she met Warren, one of her best friends, whom she came to know so well, she could identify his cough through the hospital walls. He was “so dang funny,” Jenny said, unafraid of joking about the death that would befall them both. Where she was a rule follower, he was a troublemaker. Once, he commandeered a hospital floor scrubber, waving at patients in their rooms as he drove past. Another time, he managed to procure a bootleg copy of The Avengers. Stuck in the hospital over the film’s opening weekend, he and the other CF patients organized a movie night. James brought his Xbox to play the bootleg DVD. Heather (“the biggest Swiftie”) and Angie (“gorgeous, tall blonde”) joined too. They found a waiting room with a TV, and the nurses passed around microwave popcorn.
Jenny and her friends made sure to sit several feet apart. People with cystic fibrosis have had to practice social distancing since long before COVID, because they are considered a danger to one another. Their lungs harbor destructive and often antibiotic-resistant bacteria that can become impossible to uproot once established. Certain names are spoken with an air of doom: Burkholderia cepacia, Pseudomonas aeruginosa. When doctors in the 1990s realized that people with CF were infecting and killing one another by simply gathering, they stopped allowing patients to go within several feet of one another unmasked. Camps for children with cystic fibrosis, which Jenny still remembers fondly, were all shut down. In the hospital, she once again found a community in the disease that was taking over her life. But many of those friendships ended too soon: Of the five people at the Avengers movie night, Jenny is the only one alive today. Warren, James, Heather, and Angie have all died.
As Jenny struggled with her health, the new reality of chronic illness took a toll on her marriage. She and her husband eventually divorced. After a particularly harrowing hospitalization in 2012, her doctors encouraged her to stop working and go on disability. Something in her life had to give, they told her, or it would be her body. Her disease and her daughter became her whole world.
Even as a young child, Morgan could sense when her mom was heading toward another exacerbation. If she noticed that Jenny was more tired than usual or coughing more than usual, she began to dread their coming separation. When she was 3 years old, she asked, “Do all mommies live in the hospital sometimes?” When she was 6, after Warren’s death, she asked, “Can you die from CF?” She understood that their existence together was fragile.
Jenny answered truthfully: Yes. But she assured her daughter that she was taking care of herself as best she could. Still, she made plans for what was probably inevitable. If she died, her daughter would live with her aunt and uncle. If she died, she wanted a funeral just like Warren’s, with music, candy, and an open mic for everyone to share their favorite memories.
A cure for cystic fibrosis had supposedly been imminent since 1989, when Jenny turned 2. That year, scientists identified the recessive gene behind cystic fibrosis, which encodes a protein called CFTR that controls the flow of salt and water. The discovery seemed so explosive that a Reuters reporter rushed to publish the scoop more than two weeks before the scientific papers were due to come out; two press conferences followed.
In the decades after, however, researchers came to understand the wide gulf between identifying a genetic problem and knowing how to solve it. Early attempts in the ’90s at using gene therapy to fix mutations failed again and again, both for CF and for other genetic conditions that once seemed tantalizingly close to a cure.
Then, CF researchers changed tack: Instead of correcting the gene, why not correct the mutated protein itself with small fixer molecules? This had never been done before—with any disease—but the nonprofit Cystic Fibrosis Foundation deemed the strategy promising enough to strike an unusual venture-philanthropy agreement with a company that would attempt it, which was eventually bought by Vertex Pharmaceuticals. The foundation funded the research in return for a share of the revenue.
The move paid off. In 2012, Vertex released a drug called Kalydeco that worked stunningly well—improving lung function and erasing many symptoms in the small group of CF patients who could take it. That was the catch: The FDA approved Kalydeco only for the roughly 4 percent of people with CF who carried a rare and specific mutation. Still, it provided a jolt of optimism. Kalydeco was the first drug ever tailored to a person’s inherited genetic mutation, and the breakthrough portended a new age of “personalized medicine.” It also inspired other patient-advocacy groups to copy the venture-philanthropy model. In 2014, the Cystic Fibrosis Foundation sold the rights to royalties from Kalydeco and future Vertex CF drugs for $3.3 billion, which it could invest in new research.
After Kalydeco, the next CF mutation to target was obvious. About 1,700 unique mutations have been found in people with CF, but some 90 percent of patients—including Jenny—carry at least one copy of a mutation, known as F508del, that leaves their protein channels too seriously distorted for Kalydeco alone to correct. Fixing this shape would be a much bigger task. In 2013, Jenny joined the clinical trial for a two-drug combination from Vertex, made up of Kalydeco plus a second fixer molecule. It failed to especially improve her symptoms, though it did work enough to stabilize her falling lung function. “It seemed to push pause,” she said. She stopped getting sicker, but she was still sick. The research went on.
A few years later, word began spreading of a forthcoming three-drug combination from Vertex. In clinical trials, neither patients nor doctors are told who is on the placebo and who is on the experimental drug. But in this trial, everyone could tell. The triple combo made patients’ lung function jump by a shocking 10 percentage points. Overnight, they woke up smelling for the first time the distinctive scent of their home. They could even taste their sweat becoming less salty. This was Trikafta.
In the fall of 2019, Trikafta was approved by the FDA just 10 days before a large annual gathering of CF experts in Nashville. Doctors who attended told me the atmosphere was electric. Jenny happened to be there to speak on an unrelated panel, and she remembers seeing the geneticist Francis Collins walk onstage with a guitar. Collins is best known as the longtime director of the National Institutes of Health, where he oversaw the sequencing of the human genome in the ’90s (he has since retired from the NIH). But he had made his name in 1989 as one of the scientists who discovered the gene for cystic fibrosis.
In those long years when progress was halting, Collins, who is also an amateur musician, wrote a song to inspire a gathering of CF researchers. He sang “Dare to Dream” again that day in Nashville, his baritone raspier with age. When he got to the verse that he had rewritten for this occasion—“That triple treatment has taken 30 years”—cheers broke out in the convention center. In the crowd were people who had waited their whole career, even their whole life, for this moment. We dare to dream, dare to dream. As they swayed to the music, perhaps no one quite understood the magnitude and velocity of the change to come.
Jenny received her first box of Trikafta on November 17, 2019, at the end of yet another two-week hospital stay. She had gotten sick again in Nashville. Actually, she had been fighting off a cold before she left, and despite assiduously staying in her hotel room to keep up her treatment routine, she felt an infection settling into her lungs. At the conference, she heard a lot about Trikafta, but she didn’t expect to get it so quickly. CF centers were being inundated with calls from patients asking for the new drug.
In the hospital in Utah, she recorded a video that she sent to her sister with CF, Teresa, who now lived in Ohio. She is sitting on her hospital bed. “My Trikafta is here,” she says, her voice shaking and her eyes tearing up. The miracle drug she had been promised her whole life was now in her hands.
Teresa was also able to start the drug not long after. For her, Trikafta’s impact was immediate and unmistakable. The Purge started on the drive back from the doctor’s visit where she took the first dose. The mucus coming up was so thin that she was confused; it was nothing like the sticky gunk she’d had to work so hard to cough up. A month later, she went back for a sweat test, and her salt level was normal. Based on the results, you would not know she had cystic fibrosis.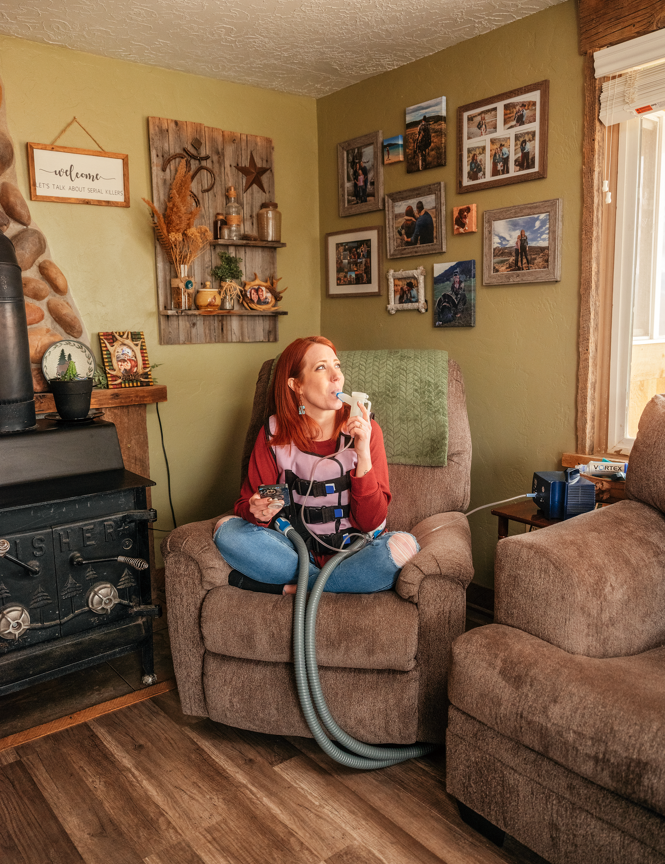
“I think of it like, ‘Oh, back when I used to have CF,’ ” Teresa said on a recent call with Jenny and me. “I don’t feel like I have CF. I feel completely normal.” She has been able to stop using her vest and inhaled medications, freeing up that time for her adopted children and the farm where she lives with her family. Before Trikafta, every small exertion was a negotiation with her lungs. Should she go upstairs? How many breaths would that take? Now she’s running around milking the goats, trimming their hooves, throwing 30 bales of hay into the barn.
On that same call, the sisters got to talking about an upcoming trip to see their grandmother, and Teresa asked Jenny a question that would have been inconceivable before Trikafta: Could they stay in the same hotel room? To avoid infecting each other with the bacteria in their lungs, the two had not shared a room since Teresa left Utah 15 years earlier. At family gatherings, they kept their distance. They didn’t even touch the same serving utensils, sending their partners to get their food. Now, Jenny told her sister, “I would totally stay in the same hotel room.”
When Jenny started Trikafta, it took her longer than it took Teresa to notice much change. She didn’t have the dramatic capital-P Purge because, she thinks, the hospitalization had already temporarily cleared her lungs. But two months after she started the drug, when a snowstorm blanketed their town, her family drove out to their favorite sledding hill. Jenny had never liked sledding; she would stand in the cold while everyone else ran around having fun, their easy breaths turning into white puffs in the air. This time, her nephew called out and she jogged over.
It wasn’t until she got to him that she realized she had jogged up—all the way to the top of the hill. “I don’t run, and I don’t climb hills. And I just ran up a hill and felt super fine,” she says in a video she took right after. “I’m going to see if I can do it again. Ready?”
“Yes,” her daughter, Morgan, answers next to her. They take off. “Mom!” Morgan shouts a few seconds later, as the distance between them grows larger. “You’re beating me, Mom!” At the top of the hill, Jenny looks back to see Morgan still catching up.
Jenny went down the hill and ran back up again, simply to prove that she could. “At one point, I just plopped up here on my bum and cried,” she told me during my visit in October, pointing to the spot on the hill where it had all hit her. In front of us, big gray mountains jutted into the blue sky. The sledding hill, she admitted, did not look that impressive. But for all of Morgan’s life, Jenny had been on the sidelines. She’d watch as Morgan swam in the lake or rode her bike, her low-grade fever making her too tired to join. That day on the hill, they finally ran together.
From there, Jenny began noticing changes in her body, big and small. The tips of her fingers, which had always been slightly swollen and round—a sign of low oxygen—thinned out as her lungs improved. She didn’t need as many enzyme pills to digest her meals. Her chronic cough disappeared. She hadn’t realized how much she had always suppressed her laughter to avoid triggering her cough. Now she can laugh—big belly laughs that match the warmth of her personality. “Oh my gosh, my laugh drives her crazy,” she told me in the car, laughing, after picking up Morgan from school. “That’s because you laugh at stuff that’s not funny,” her daughter shot back. Jenny laughed again.
Trikafta had effects that even doctors did not anticipate. In the months after the drugs became widely available, some patients unexpectedly got pregnant; the drug that thins lung mucus, it turns out, also thins cervical mucus. Then, patients started trying to get pregnant. The drug made many people with CF feel so healthy that they no longer worried about the physical toll of pregnancy and parenthood or the agony of leaving behind young children. Doctors began speaking of a Trikafta baby boom.
Doors opened to other once-impossible futures. A 22-year-old told me he decided to train as an aircraft mechanic, a job that would have been far too physically demanding when he was being hospitalized multiple times a year. One woman started dating. “I don’t want to fall in love with somebody, knowing that I’m not going to be around very long,” she had thought. Now she and her boyfriend have been together for four years. A father who was being evaluated for a lung transplant before Trikafta felt healthy enough to spend the summer of 2020 tearing down and rebuilding his family’s deck, and now expects his CF lungs to see him through graduations and grandkids.
Trikafta is a lifelong medication, and it is not meant to undo organ damage that has already occurred. But the earlier treatment begins, the healthier one stays. A handful of pregnant women have now used Trikafta to treat their unborn children with cystic fibrosis. Last fall, I corresponded with one such expecting mother, who does not have CF but whose son was diagnosed by genetic testing. She started Trikafta at 26 weeks. When her son was born in October, his lungs and pancreas were perfectly healthy.
Officially, Trikafta is approved in the U.S. for patients as young as 2. Unofficially, some parents give their newborns Trikafta, either indirectly through breast milk or directly by grinding up the pills into tiny doses. So long as they stay on the medication, these children may never experience any of the physical ravages of the disease. Recently, Make-A-Wish announced that children with CF would no longer automatically be eligible for the program, because “life-changing advances” had radically improved the outlook for them.
CF centers these days are unusually quiet. Fewer patients need once-routine weeks-long hospitalizations. Instead of thinking about lung function, more and more are worrying about the maladies that come with middle and old age—colon cancer, high cholesterol, heart disease. Obesity has been a confounding new issue. Before Trikafta, patients were usually underweight, and they were told to cram as many calories in as possible, by whatever means possible. Every additional pound was a small victory. One patient described microwaving pints of Ben & Jerry’s to drink mixed with heavy cream; when even that failed to make her gain weight, she got a feeding tube. Now people on Trikafta worry about getting too many calories.
In February, Vertex announced the results of a clinical trial for a next-generation triple-combination therapy, which may be even more effective than Trikafta. All of these changes have made for an existential moment for doctors, too: The disease they were trained to treat is no longer the disease most of their patients have.
Doctors told me they could think of only one other comparable breakthrough in recent memory: the arrival of powerful HIV drugs in the 1990s. Like Trikafta, those drugs were not a cure, but they transformed AIDS from a terminal illness into a manageable chronic one. Young men got up from their deathbed, newly strong and hale. AIDS hospices emptied—and then went bankrupt.
This was a remarkable turn of events. But it elicited a complicated mix of emotions, not all of them joyful. Some patients who were no longer dying grew depressed, anxious, and even suicidal at the thought of living. This phenomenon became known as “Lazarus syndrome.”
Death is an end, after all. Life comes with problems: Patients who spent lavishly during what were supposed to be their last days now had no money to live on. Those who stayed with a lover in sickness found that they could not actually stand them in health. They fretted about insurance and paperwork and chores, everyday annoyances that would no longer be obliterated by imminent death. In 1996, the writer Andrew Sullivan, who is HIV-positive, described life after the advent of the HIV drugs in his essay “When Plagues End”:
When you have spent several years girding yourself for the possibility of death, it is not so easy to gird yourself instead for the possibility of life. What you expect to greet with the euphoria of victory comes instead like the slow withdrawal of an excuse. And you resist it.
The intensity with which you had learned to approach each day turns into a banality, a banality that refuses to understand or even appreciate the experience you have just gone through.
For some HIV patients, their reversal of fortune seemed unreal. “He doesn’t trust what’s happening to him,” one doctor said about a patient who had made a dramatic recovery, yet found himself in psychological distress.
Doubts like these crept into the minds of many people on Trikafta, too. What if the new drug stopped working? Or had horrible side effects? Or stopped being covered by insurance? Trikafta’s sticker price is more than $300,000 a year. Insurance typically covers most of that cost—minus what can be significant co-pays and deductibles—and Vertex offers co-pay assistance. But patients’ lives ultimately depend on decisions made by nameless bureaucrats in rooms far away: Insurance plans can suddenly change what they cover, and in 2022, Vertex announced that it would substantially reduce its financial assistance.
A 43-year-old woman I interviewed asked not to be named, because she feared that speaking about her improved health would cause her to lose disability benefits, which would also get her kicked off the government insurance that pays for Trikafta. Her health has not improved as dramatically as others’ has, and she still has frequent infections and occasional bleeding in her lungs. If she returns to work but her health declines, it could take a long time to get back on disability—time she would have to go without Trikafta. She would also need a job with health insurance good enough to cover the expensive drug—but could she even get one as a 40-something with no recent employment history?
For other patients, new health granted new independence, which could be scary too. As a child, Patrick Allen Brown was sick enough to miss long stretches of school. His parents didn’t expect him to do chores, let alone support himself with a job one day. So much of his life was spent in the hospital that movies became his way of understanding the outside world. In his teens and 20s, he drank heavily.
After Trikafta restored Brown’s physical health, he was no longer a chronically ill adult who lived with his parents. He was a pretty healthy adult who still lived with his parents. He was 32, and hadn’t finished college. Now he had to budget, commit to a career. He decided to get sober. When one of his parents needed back surgery recently, their roles flipped: He became the caretaker. Brown has now graduated from culinary school and found work as a chef, but he feels as if he is still catching up to his peers.
The great blossoming of possibilities on Trikafta also dredged up regret about decisions too late to undo. Kara Hansen, 41, has a daughter who was adopted, and she had always wanted another child. But in 2016, she had to be repeatedly hospitalized: in April, then again in May, July, and August. She gave up on having a second child—how could she, if she couldn’t even guarantee living for the daughter she already had? Then, in 2018, she joined the original trial for Trikafta, becoming one of the first people in the world to experience its miraculous effects. If she had known her health would improve so dramatically and hold steady six years on, she would have tried to get pregnant, but she feels like it’s too late now. To plan for such a miracle would have been foolish, but to live in its unexpected aftermath can still be painful.
After a year on Trikafta, Jenny told Teresa something that she acknowledged sounded “insane” but that her sister understood immediately: “To no longer be actively dying kind of sucks,” she said. The certainty of dying young, she realized, had been a security blanket. She’d never worried about retirement, menopause, or the loneliness of outliving a parent or a partner.
Cystic fibrosis had defined her adult life. Now what? For so long, she’d just been trying to see her daughter graduate from high school. Now she faced seeing Morgan go off and live her own life. What then? Jenny had become active in patient advocacy, and soon after the start of the pandemic, she volunteered to moderate an online patient forum on mental health for her CF center in Utah. It went so well that her longtime social worker at the center felt compelled to give some career advice: Try social work.
[From the October 2022 issue: Sarah Zhang on why so many kids need glasses now]
Jenny enrolled in an online master’s program in 2022, and this past fall she chose a practicum with a hospice agency. Having watched the death of so many friends and contemplated her own, she felt prepared to shepherd people through the sadness and awkwardness and even humor that accompany the end of life. She understood, too, the small dignities that mean the world when your body is no longer up to the task of living. One hospice patient, she noticed, often had trouble understanding conversations because his hearing aids were never charged correctly. She got the situation fixed, and on a recent visit, he wanted to listen to music, playing for her the favorite songs of his youth. On another man’s shelf, she recognized a birding book, and she made plans for a window feeder to bring birds to him.
Jenny doesn’t share the details of her life with patients, but in their experiences with death, she has seen her own refracted. One hospice patient, a devout elderly woman, was estranged from her adult son, who no longer believed. Jenny herself grew up religious—Mormon, in her case—but she is not anymore. Her family is still Mormon, as is virtually everyone in the town she has lived in since childhood, which has 3,500 people, several Mormon churches, and a Mormon temple. She is liberal, whereas most of her relatives voted for Donald Trump.
Still, Jenny has made a point of staying close to her large, tight-knit family. Knowing she would die young had long ago clarified that she wanted to leave with no regrets, no grudges, and no words left unsaid to the people she loved. In the foothills outside town one day, she pointed in the direction of her house, her brother’s house, her mom’s house, her dad and stepmom’s house, all minutes away from one another.
Although Trikafta looks to be a very safe drug for most people, it does have side effects. It can cause cataracts as well as liver injury. More perplexing, Trikafta may affect the brain.
For Jenny, starting Trikafta coincided with a wave of intense insomnia, brain fog, and anxiety. For months, she could sleep only two or three hours a night. She’d lose her phone and find it in the freezer. Her lungs were so much healthier, but her brain was going haywire. Soon, she realized that other CF patients had begun sharing stories online of depression, anger, or suicidal thoughts that emerged at the same time they started taking Trikafta.
Doctors sometimes chalked up these symptoms to the existential unease of no longer dying, or the fear and isolation everyone felt in the early days of the pandemic. But Jenny’s doctor took the side effects she reported seriously enough to suggest that she halve her Trikafta dose, and soon after, they subsided. (Some of her CF symptoms did return, but they were muted enough that she could pare down her regimen of treatments.)
The link between Trikafta and these symptoms in the brain is still not fully proven or understood. “We’ve done an in-depth analysis of the preclinical data, clinical data, and real-world-evidence data, and we don’t find any causal relationship,” Fred Van Goor, a vice president and the head of CF research at Vertex, told me in January. And an analysis co-authored by the company’s scientists last year found similar rates of depression and suicidality in CF patients with or without Trikafta. But in November, a group of scientists published a review arguing that the possible neuropsychiatric effects of Trikafta deserved a “serious research effort.” The protein behind CF is found in cells throughout the body, including the brain. Trikafta could be acting on the brain directly, the authors hypothesized, or it could be acting indirectly via changes to inflammation throughout the body or specifically in the gut. The drug may affect different subsets of patients differently, says Anna Georgiopoulos, a psychiatrist at Massachusetts General Hospital who co-authored the review. She believes that neuropsychiatric side effects afflict only a “small minority” of people on Trikafta, but says that studies are needed to know exactly how many.
In the meantime, some patients have quit Trikafta altogether, their neuropsychiatric symptoms too debilitating even on a lower dose. “Physically I was feeling the best I’ve ever felt,” says Aimee Lecointre of her time on the drug, but mentally, “I felt on the verge of a panic attack almost every day.” The contradiction confused her: How could she be so anxious and depressed when her health was getting so much better? When she finally decided to try stopping Trikafta, the nervous energy that had filled her body all day long dissipated. But her CF symptoms came back. During our phone conversation, she paused every few minutes to cough.
She and Jenny have known each other for years, going back to their mutual hospitalizations. The three of us were supposed to meet over apple-cider floats when I was in Utah, but Lecointre had health issues come up at the last minute, the kind of disruption that happens all the time for people with a chronic illness. For a while, her Instagram feed filled with people on Trikafta whose lives were transforming while hers stayed the same; she had to delete social media from her phone. She still feels sad, sometimes, that Trikafta didn’t work out for her. But she was able to go back to one of Vertex’s two-drug combos, and although it is less effective than Trikafta, she feels so much better. There is more to cope with, but the coping is easier.
For another group of CF patients, Trikafta simply does not work. About 10 percent lack the F508del mutation that the triple combination was specifically designed to fix. Over time, though, scientists have found that some less common mutations are similar enough to F508del that those who carry them still benefit from Trikafta. And in late 2020, word got out that the FDA would soon approve the drug for additional mutations.
Gina Ruiz remembers waiting and waiting for the list of new mutations that fall. She had spent the past year watching her peers on Trikafta be handed what she thought of as a “reverse Uno card”—reverse weight loss, reverse lung decline, reverse CF—while her own health continued to worsen. She was sitting in a car when she saw the list, and she scrolled through the 177 new mutations hoping to find hers. She was crushed when she did not. Ruiz and most people in the 10 percent have mutations that leave their CFTR protein too garbled or incomplete to correct with any combination of fixer molecules. Treating these mutations will require a different strategy altogether.
The Cystic Fibrosis Foundation continues to fund research into a cure for all, and scientists, including those at Vertex, are once again exploring genetic therapies, applying the lessons of past failures. But a genetic-therapy breakthrough specific to CF is still years, if not decades, away. After Vertex created that first drug for the 4 percent, the path toward Trikafta was clear. After Trikafta, terra incognita.
Ruiz is wary of getting her hopes up again. At age 29, she can no longer work. She lives with her parents. Her lung function has fallen to 30 percent. And in December, her weight reached a new low of 89 pounds. “I went to Target last night and I was beyond exhausted,” she told me the following month. Her knees hurt too, another complication of CF. As she’s watched her peers on Trikafta get married and chase after toddlers, her own world has shrunk. Halfway through the store, she got so tired that she had to rest in a chair in the home-goods section before she could go on.
Other patients with rare mutations told me the CF communities they once relied on for support have become quiet, as the 90 percent have gotten on with their lives. “It’s extremely isolating,” says Steph Hansen, who was steeling herself for another hospitalization when we spoke in January. She describes it as a one-two punch: Her health is no better, yet she has lost the community that once buoyed her. She’s connected with a handful of other patients who can’t take Trikafta, but CF is already a rare disease, and they are the rarest of the rare.
The F508del mutation is most common in people of European ancestry, so people with mutations ineligible for Trikafta in the U.S. are disproportionately Black or Latino. Globally, the proportion of people ineligible is higher in Latin America, Asia, and Africa, where diagnosis and treatment for CF also lag. In most developing countries, even eligible patients cannot get Trikafta—because Vertex currently does not sell its expensive drug outside a few dozen countries, concentrated in Europe and the English-speaking world. (Vertex says it has a pilot program that “provides Trikafta at no cost to people with CF in certain lower income countries.”) Its patents also block other companies from making a cheaper generic version. In early 2023, activists asked four countries to revoke or suspend patents for Trikafta in a coordinated campaign. One of the countries was India, where The New York Times wrote about a father named Seshagiri Buddana. His son would have been able to take Trikafta if he lived in the U.S., but he died in December 2022 one day before he would have turned 9.
All of this weighs on Jenny. What makes her different from those who have died, other than the luck of being born at the right time, in the right place, with the right mutations?
Two days after my visit to Utah, Jenny’s father, Tom, had a heart attack while chopping firewood. He felt short of breath, and a trip to the hospital revealed that his major arteries were 90 percent blocked.
When Jenny texted me the news, she said she had been replaying our recent conversations about life and death. She was glad to feel, upon learning her father might die, that nothing between the two of them was left unsaid or unresolved. I thought of what Tom had told me in his living room. Before we had gone over to his house that day, Jenny had warned me that her dad was a jokester, not a man prone to earnest reflection. But when the conversation shifted to the impact of Trikafta, he turned to me, completely serious. “I was going to bury my kids. And I’m not. They get to bury me, which is the way it’s supposed to be.”
We all fell silent for a moment, as we felt the weight he had been carrying all those years. After burying his eldest daughter at 14, Tom could no longer watch movies in which children die. In Jenny’s years of sickness, he had often driven her two hours to the hospital in Salt Lake City, but he rarely set foot inside. Hospitals are places where people go to be born or to die, he’d say, and all my children have already been born.
After his heart attack, Tom needed an emergency quintuple-bypass surgery. He did well, and came home to recover. He spent the time rethinking his priorities. Just before falling ill, he had skipped a family outing to an amusement park to work. Now he regretted it. He’s become more open about his emotions; still a jokester, he’s taken to saying that his heart has been opened in more ways than one since the surgery.
It’s interesting, Jenny says. Her father has lived a longer and very different life from her own, but she recognizes what he is going through. People die from this, he started saying. I could have died from this. He got close enough to see death’s shadow, only to be pulled back to a life whose familiarity suddenly felt unfamiliar. What would he do with his unexpected life? “Hey,” Jenny told her dad. “I get it.”
This article appears in the April 2024 print edition with the headline “After the Miracle.”
What's Your Reaction?




















